Déficit en G6PD

INTRODUCTION/GÉNÉRALITÉ
Votre texte ici
HISTORIQUE
Votre texte ici
PHYSIOPATHOLOGIE
- L’enzyme G6PD intervient à la première étape de la voie des pentoses‐phosphates: cette voie alternative de la glycolyse, permet de produire du NADPH à partir de l’oxydation du glucose-6-phosphate.
- Le NADPH est le co-enzyme de la glutathion réductase qui permet la production du glutathion réduit (GSH) molécule indispensable pour lutter contre certaines oxydations. La G6PD est donc indispensable au contrôle du stress oxydatif.
- Le globule rouge est une cellule particulièrement exposée aux radicaux oxydants du fait de l’abondance de l’oxygène et du fer dans son cytoplasme. Le globule rouge étant dénué de noyau, il ne produit ni ARNs messagers ni protéines et ne peut donc renouveler son stock enzymatique. La voie des pentoses phosphates est la seule voie de production du NADPH dans les globules rouges contrairement aux autres cellules qui peuvent en produire par la voie de l'enzyme malique. Enfin la G6PD est une protéine assez instable et cela aboutit à une activité G6PD quasi nulle dans les globules rouges les plus âgés ; à contrario en cas de rajeunissement de la population des globules rouges, l’activité G6PD augmente notablement.
- Si le déficit enzymatique en G6PD est le plus souvent asymptomatique il peut entraîner une hémolyse aiguë lors de l’exposition à un stress oxydatif en particulier à certains produits pro-oxydants alimentaires ou médicamenteux.1
ÉPIDÉMIOLOGIE
- Le déficit en G6PD figure parmi les affections génétiques les plus répandues dans le monde.
- Il touche plus de 400 millions de personnes, essentiellement originaires d'Afrique, d'Asie, d’Inde, du Moyen-Orient et du Bassin Méditerranéen. La grande majorité d’entre elles restera asymptomatique.
- Le gène codant pour la G6PD est porté par le chromosome X et les sujets symptomatiques sont majoritairement de sexe masculin. Ils sont dits hémizygotes.
- Les garçons hémizygotes et les rares sujets féminins homozygotes sont déficitaires.
- Les sujets féminins hétérozygotes sont le plus souvent «conductrices» sans être malades. Cependant, du fait de l’inactivation aléatoire d’un des 2 chromosomes X, un seul des 2 allèles est actif dans chaque cellule. En conséquence, les globules rouges d’une femme hétérozygote forment une mosaïque : une partie est déficiente, l’autre non et le plus souvent l’activité résiduelle est aux environs de 50% de la normale. L’inactivation préférentielle de l’X vers l’allèle déficitaire ou l’allèle normal peut conduire chez l’hétérozygote à un déficit franc ou au contraire une activité G6PD apparemment normale.
- Les mutants G6PD déficitaires sont plus répandus dans les régions du monde où a existé une endémie palustre car ils confèrent au sujet porteur un degré de résistance vis à vis de ce parasite. Cette protection a été montrée dans des populations africaines où les personnes déficitaires ont un risque de développer une forme sévère de paludisme à Plasmodium falciparum diminué d’environ 50%.
- Bien qu’un grand nombre de variants soit décrit, il existe, en règle, dans chaque zone géographique un variant déficitaire dominant : variant A- pour les populations issues d'Afrique, variant B- (appelé aussi variant Med) pour celles du Bassin Méditerranéen et du Moyen-Orient, variants Mahidol et Viangchan en Asie du Sud Est, variants Canton et Kaiping en Chine… Tous ces variants peuvent entraîner un accident hémolytique aigu en cas de stress oxydatif impliquant le rôle protecteur du glutathion.
- Les deux variants déficitaires les plus fréquemment retrouvés en France sont le variant «A- » et « B- ou Med ». Ils représentent 80% des variants détectés (60% A-, 22% B-). Le variant «A- » est très répandu chez les sujets originaires d’Afrique sub-saharienne et le variant « B- ou Med », retrouvé chez les personnes originaires du bassin méditerranéen.
- Les données OMS de 1989 estimaient le pourcentage des hommes hémizygotes déficitaires en G6PD à 0,39% (soit environ 120 000 déficitaires) en France métropolitaine et à 12% en Martinique et en Guadeloupe.1
CLASSIFICATION
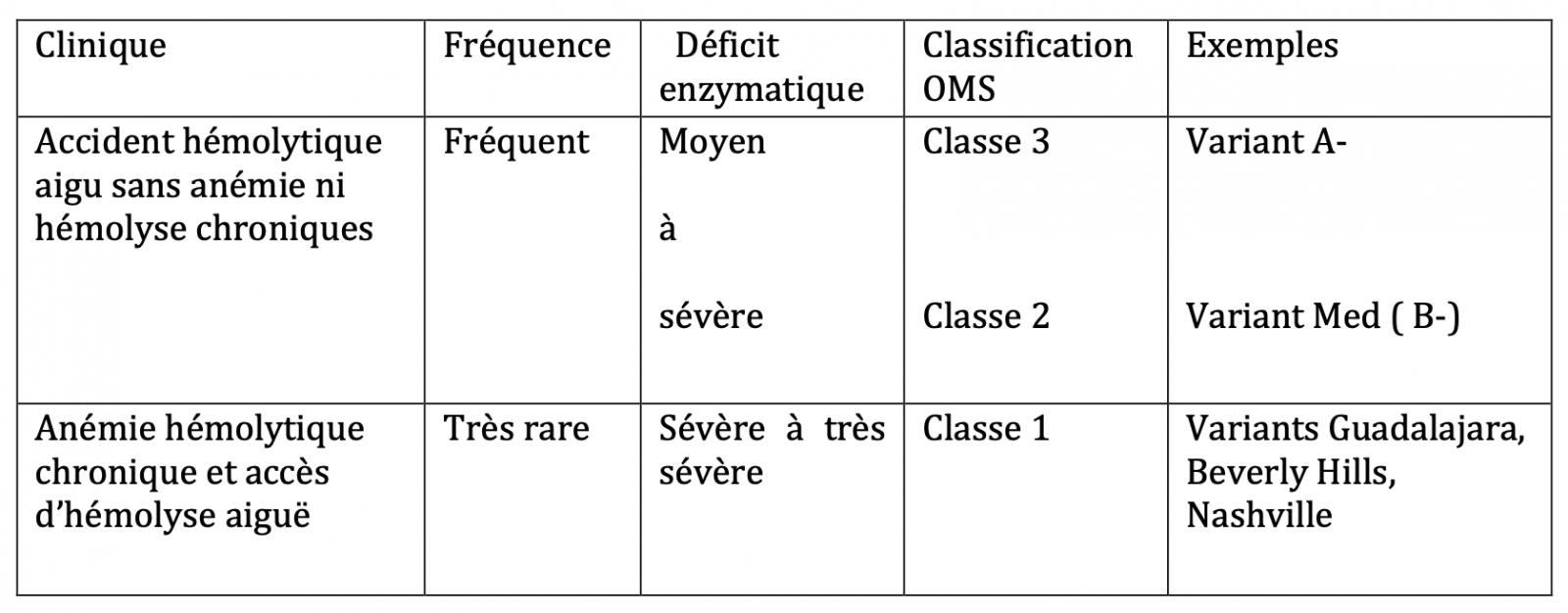
Plus de 200 variants G6PD résultant majoritairement de mutations faux-sens ont été rapportés. Leur conséquence est très variable, principalement fonction de l’importance de la baisse de l’activité enzymatique.
L’OMS a classifié les variants G6PD en 5 catégories (3 déficitaires) selon la mesure de l’activité G6PD :
- Dans la classe 1 le déficit est très sévère (<10%, le plus souvent <1% ou indosable) et, fait particulier, il détermine une anémie hémolytique chronique.
- L’activité est abaissée dans la classe 3 (60% à 10 % de la normale) et la classe 2 (activité < à 10 %). Les classes 2 et 3 sont les classes déficitaires les plus fréquentes, entraînant les accès d’hémolyse aiguë liés à un stress oxydatif caractéristiques du déficit en G6PD.
- Elle est normale dans la classe 4 qui regroupe les deux variants les plus fréquents à activité « normale » A et B (qui diffèrent par un polymorphisme neutre) et les autres variants neutres de ces protéines.
- L’activité est supérieure à la normale dans le type 5.
EXAMEN CLINIQUE
- Les patients déficitaires sont le plus souvent asymptomatiques.
- Le diagnostic de déficit en G6PD est évoqué sur des symptômes cliniques ou dépisté chez un sujet asymptomatique par exemple dans le cadre d’un bilan familial, avant l’administration d’un médicament oxydant contre-indiqué ou encore dans le cadre du bilan d’une autre maladie du globule rouge (drépanocytose, thalassémie….).
- Les deux principales présentations cliniques sont l’ictère néonatal et l’accident hémolytique aigu secondaire à la prise de médicaments oxydants, de fèves ou contemporain d’un épisode fébrile. La présence d’une anémie hémolytique chronique est exceptionnelle et caractérise les déficits de classe 1.
A) Ictère néonatal
- L’ictère, symptôme d’une hyperbilirubinémie est très fréquent, chez le nouveau-né. Il apparaît entre J2 et J3, progresse jusqu’à J5-J7 puis régresse et disparaît avant J15. Il témoigne de l’adaptation physiologique du métabolisme de la bilirubine caractérisé par un déséquilibre entre production importante (dégradation de l’hème) et élimination réduite de la bilirubine (immaturité hépatique et digestive). Cet ictère est à bilirubine non conjuguée ou libre dans 99% des cas.1 La première quinzaine de vie représente le temps nécessaire à l’adaptation pour que l’équilibre production/élimination de la bilirubine soit atteint.
- En cas de déficit en G6PD, cette évolution peut être perturbée du fait de l’hémolyse induite. La bilirubine produite en excès peut alors s’accumuler au-delà des valeurs normales (physiologiques) pour l’âge post-natal. La bilirubine est potentiellement neurotoxique avec une affinité plus particulière pour les noyaux gris centraux et les centres auditifs.1 Cette liaison peut être responsable d’une encéphalopathie aiguë réversible ou chronique et décrite alors sous le nom d’ictère nucléaire. Ce tableau qui reste rare associe des séquelles motrices (coordination des mouvements, paralysie du regard) et une surdité centrale. Si le risque de complications neurologiques est décrit pour un taux de bilirubine circulant > 340 μmol/ l, 3/4 des ictères nucléaires décrits font suite à une bilirubinémie > 520 μmol/l. Néanmoins l’origine hémolytique de l’hyperbilirubinémie augmente le risque de neuro-toxicité de celle-ci. Le déficit en G6PD représente un quart à un tiers des étiologies identifiées dans toutes les cohortes d’hyperbilirubinémies sévères ou des indications d’exsanguino-transfusion.1 1 1 1
- Le déficit en G6PD est le plus souvent asymptomatique en période néonatale mais dans la population déficitaire l’ictère est plus fréquent et plus sévère et représente la principale manifestation clinique 1 1 qui survient toujours dans les 15 premiers jours de vie.1 1 1 1 Contrairement à ce qui est observé plus tard dans la vie, l’hémolyse n’est généralement pas déclenchée par un stress oxydatif. Néanmoins, l’interrogatoire doit toujours chercher à identifier une exposition alimentaire ou médicamenteuse en particulier via le lait maternel.1 1 1 1 1
Deux types de présentation clinique révélant un déficit en G6PD existent: 1 1
- une hyperbilirubinémie aiguë et sévère correspondant à une hémolyse aiguë entre J4 et J10. Les bilirubinémies sont très élevées avec risque de toxicité neurologique.
- une hyperbilirubinémie précoce (dans les 24 à 36 premières heures de vie), volontiers prolongée sur la première semaine de vie, non expliquée par une cause immunologique et sans hémolyse patente à la première NFS.
- Du fait de la transmission liée à l’X, les garçons sont plus souvent affectés que les filles mais celles-ci peuvent développer un ictère grave.1 1 1 1
- Le diagnostic de déficit en G6PD est posé sur demande de dosage d’activité G6PD devant tout ictère sévère ou persistant non immunologique (test direct à l’antiglobuline et élution à anti-A ou B selon le contexte d’incompatibilité négatif) chez des nouveau-nés (garçon ou fille) de parents originaires de zones à forte prévalence. Les valeurs de référence de l’activité G6PD à la naissance sont plus élevées que chez l’enfant de plus de 6 mois.1 Le diagnostic différentiel dans le cadre des hyperbilirubinémies sévères à bilirubine libre est principalement l’incompatibilité fœto- maternelle ABO, de survenue généralement plus précoce (J1-J3) et les autres hémolyses constitutionnelles (maladies de membrane érythrocytaire, déficit en pyruvate kinase, ..). Les nouveau-nés déficitaires en G6PD qui sont également porteurs d’un « syndrome de Gilbert » (anomalie génétique familiale de la conjugaison de la bilirubine) ont un risque majoré d’ictère néonatal.1 1 1 1
B) Hémolyse aiguë après exposition alimentaire ou médicamenteuse ou lors d’une infection
- L’hémolyse aiguë est secondaire à la prise de médicaments (ou produits) oxydants ou de fèves (favisme), ou contemporaine d’un épisode infectieux. Elle est dans ce dernier cas d’intensité souvent plus modérée.
- Elle survient dans les 24 heures à 3 jours suivant la prise de l’agent oxydant, de manière variable, même pour un individu déficitaire donné.
- De nombreux facteurs (dose du médicament, type de déficit, âge, co-morbidités) peuvent moduler la gravité de l’anémie.
- L’anémie se majore jusqu’à 7–8 jours après l’arrêt du médicament causal et se répare en règle à partir de 8-10 jours.
- Le favisme s’accompagne de signes d’hémolyse souvent plus précoces et sévères que ceux observés après la prise de médicaments oxydants.
- L’anémie (asthénie, pâleur) peut être très aiguë et sévère en particulier chez l’enfant et nécessiter une transfusion en urgence.
- Les douleurs abdominales ou lombaires, les urines « porto » ou « coca-cola », témoignent d’une hémolyse intravasculaire avec hémoglobinurie. L’ictère apparaît secondairement.
- L’hémolyse peut se compliquer principalement chez l’adulte d’une insuffisance rénale aiguë et nécessiter une courte période de dialyse.
- D’autres circonstances (décompensation acido-cétosique d’un diabète, infarctus du myocarde) ont été rapportées comme déclenchantes d’une poussée d’hémolyse avec toutefois une fréquente exposition médicamenteuse ou une infection contemporaines.
- Les principaux diagnostics différentiels sont les anémies hémolytiques aiguës d’une autre origine (auto-immune, mécanique). Egalement d’autres affections constitutionnelles du globule rouge (certaines hémoglobines instables et les hémoglobinoses H, les rares déficits des enzymes de la synthèse ou du recyclage du glutathion) peuvent occasionner des accidents hémolytiques au cours d’un épisode infectieux ou post-médicamenteux. La maladie de Wilson, l’hémoglobinurie paroxystique nocturne sont également responsables d’épisodes hémolytiques aigus.
C) Anémie chronique des déficits de classe I
- Contrairement aux autres catégories de déficit en G6PD, les variants de classe 1 déterminent chez les garçons une anémie hémolytique chronique de sévérité variable, s’accompagnant d’une réticulocytose élevée. Ces cas sont très rares.
- L’ictère néonatal est très fréquemment présent et peut s’aggraver très rapidement. Des cas exceptionnels d’anémie fœtale ont été rapportés.
- Les complications habituelles des hémolyses chroniques (splénomégalie, ictère, lithiases biliaires, érythroblastopénie à parvovirus (érythrovirus) B19 sont rencontrées. Les poussées d’hémolyse aiguë intravasculaire en particulier lors d’épisodes fébriles exacerbent l’anémie déjà présente. Les transfusions sont le plus souvent ponctuelles, rarement itératives induisant alors une surcharge en fer.
- Le diagnostic de déficit en G6PD de classe 1 est donc évoqué devant une hémolyse chronique constitutionnelle après élimination des autres causes en particulier d’une sphérocytose héréditaire ou, dans le cadre des déficits en G6PD déjà diagnostiqués si l’hémolyse persiste à distance d’un accident hémolytique aigu.
- Les déficits de classe 1 sont décrits dans toutes les populations et plus de 70 mutations causales ont été rapportées. Les antécédents familiaux sont inconstants, les mutations pouvant survenir de novo. Les sujets féminins conductrices d’un déficit de type 1 présentent généralement un dosage de G6PD normal du fait d’une sélection érythrocytaire fortement biaisée au profit de l’allèle normal, les cellules inactivant l’allèle normal ayant une survie compétitive très diminuée. Il a été décrit chez certaines « conductrices » un affaiblissement de cette sélection et, après 50 ans, l’émergence d’une tendance hémolytique témoignant de l’apparition de cellules déficitaires. Il est donc nécessaire, d’étudier le gène de la G6PD chez les mères d’un enfant porteur d’un déficit de classe 1 et de surveiller leur dosage enzymatique après 50 ans.
EXAMENS COMPLÉMENTAIRES
A) Diagnostic biologique
1) Hémogramme et bilan d’hémolyse
- Lors d’un épisode d’hémolyse aigu, l’hémogramme retrouve une anémie parfois très sévère avec une réticulocytose élevée.
- Le diagnostic peut être évoqué sur la présence de corps de Heinz dans les globules rouges, recherchés sur le frottis par la coloration au bleu de crésyl ou au violet de méthyl ou sur des anomalies cytologiques évocatrices de déficit en G6PD (hématies mordues ou pincées, hématies fantômes ou hemighost).
- Les marqueurs d’hémolyse sont perturbés: LDH et bilirubine libre (ou indirecte) élevées et haptoglobine effondrée.
- En dehors des épisodes d’hémolyse, l’hémogramme est normal excepté chez les rares patients atteints d’un déficit de classe 1 où il montre une anémie chronique régénérative. A l’état basal, chez les déficients de classe 2 ou 3, les marqueurs habituels d’hémolyse (haptoglobine, réticulocytose, LDH, bilirubine totale et indirecte) sont normaux.
- L’hémoglobine glyquée est également normale. Sa valeur dans le diagnostic et le suivi du diabète peut être mise à défaut en cas de déficit en G6PD car sous-estimée du fait d’une diminution du pool des globules rouges les plus âgés.
2) Dosage enzymatique
Le diagnostic repose classiquement sur le dosage quantitatif spectrophotométrique de l’activité enzymatique érythrocytaire. Il s’effectue sur un prélèvement sanguin réalisé sur anticoagulant (citrate ou mieux acide citrique dextrose). Les variants déficitaires G6PD étant le plus souvent des protéines instables, il est recommandé de transférer immédiatement le tube vers un laboratoire compétent ou, si le temps doit excéder 24h, de réfrigérer le prélèvement et de le transférer entre +4 °C et +6 °C. Le dosage doit impérativement être effectué à distance d’une transfusion sanguine (au moins 3 mois).
L’interprétation du dosage est délicate :
- Ainsi, après un accès hémolytique, même fruste, le dosage de la G6PD peut être faussement normal car les globules rouges survivants après la crise sont les plus jeunes et les moins déficitaires. Une méthode pour détecter ce biais, et ceci est vrai pour toutes les mesures d’activité enzymatique érythrocytaire, consiste à demander au laboratoire d’effectuer sur le même prélèvement la mesure d’une autre activité enzymatique érythrocytaire (hexokinase ou pyruvate kinase) qui servira de contrôle interne de l’âge moyen des GR étudiés. Par exemple, une mesure G6PD normale mais avec une activité hexokinase à 2-3 fois la normale, sur un prélèvement riche en réticulocytes, est en faveur d’un déficit en G6PD. La réticulocytose est donc un élément important à prendre en compte dans l’interprétation du dosage.
- De plus, du fait de la présence du gène sur le chromosome X, l’activité enzymatique résiduelle dépend du sexe du patient. Chez les garçons hémizygotes ou les filles homozygotes le résultat est toujours franchement abaissé. Le dosage spectrophotométrique détecte également les déficits partiels (femmes hétérozygotes) mais n’est pas corrélé parfaitement au génotype du fait de la possibilité d’une inactivation cellulaire de l’X non équilibrée. En effet, chez les sujets féminins hétérozygotes le dosage est variable, de normal à franchement déficitaire.
- Enfin, on notera que chez les sujets présentant une diminution de la quantité d’hémoglobine par globule rouge ou hypochromie (thalassémie, carence martiale), l’activité enzymatique peut être artificiellement augmentée et un déficit masqué en particulier chez une femme hétérozygote.
- Des méthodes semi-quantitatives cellulaires mettant en jeu la présence de NADPH ou la réduction par l’enzyme de formazan sont appliquées pour le dépistage de masse du déficit en G6PD mais elles sont moins performantes que le dosage de référence pour le diagnostic des hétérozygotes.
- Une fois porté dans de bonnes conditions, il n’est en général pas nécessaire de contrôler le diagnostic biologique de déficit en G6PD sauf lorsque le premier dosage a été pratiqué en période néonatale ou au cours des 1ers mois de vie. Il sera alors proposé un contrôle après l’âge de 9-12 mois.
3) Diagnostic moléculaire
- L’étude moléculaire qui va identifier la mutation en cause (génotypage) est indiquée dans tous les rares cas d’anémie hémolytique chronique (déficits de classe 1) où elle confirme le diagnostic.
- Elle peut être proposée à visée diagnostique dans les autres cas de déficit (accident d’hémolyse aigu correspondant aux classes 2 et 3) quand le dosage ne permet pas le diagnostic : chez un patient déficitaire ayant reçu une transfusion récente ou très réticulocytaire et chez certains sujets féminins pour préciser leur statut homozygote ou ou hétérozygote
B) Bilan initial
- Chez un sujet symptomatique (hémolyse, ictère) le bilan initial comporte: l’hémogramme avec compte des réticulocytes, l’examen du frottis sanguin (morphologie érythrocytaire), la bilirubine totale et indirecte, la fonction rénale, le dosage G6PD, la détermination du groupe sanguin et phénotype Rh Kell, la recherche d’agglutinines irrégulières. La détermination du phénotype érythrocytaire étendu aux antigènes Jk, Fy et MNS sera secondairement demandée dans les rares cas de déficit responsable d’une anémie chronique (classe 1). Les sérologies VIH, VHC, VHB ne sont plus indiquées à titre systématique dans le suivi transfusionnel, mais font partie du bilan initial. Dans les déficits de classe I, le bilan initial comporte également une échographie abdominale de référence et une sérologie parvovirus B19.
- Chez un sujet asymptomatique, le dosage sera prescrit dans le cadre d’un dépistage familial ou avant l’administration d’un médicament contre-indiqué en cas de déficit en G6PD. Le bilan sanguin comporte l’hémogramme, les réticulocytes et le dosage G6PD
C) Bilan familial
- Dans les déficits habituels (accident hémolytique aigu sans hémolyse chronique) correspondant aux types 2 et 3, les explications sur le mode de transmission sont délivrées par le médecin pédiatre, interniste, hématologue ou lors d’une consultation de génétique. Un diagnostic prénatal n’est pas indiqué mais un dépistage du déficit à la naissance est préconisé.
- Hors contexte néonatal, la pratique du dosage enzymatique est proposée aux frères et oncles maternels du propositus masculin. La mère du propositus est au minimum hétérozygote. Pour les autres membres de la famille de sexe féminin le dosage n’est pas systématiquement proposé.
- Si le propositus symptomatique est féminin (hétérozygote avec biais d’inactivation ou plus rarement sujet homozygote) le bilan familial sera plus large incluant en particulier le père du propositus et ses sœurs.
- Dans le cadre particulier des déficits de classe 1, une consultation de conseil génétique est indiquée. Elle précisera le statut maternel (conductrice, mutation de novo) et dans les formes sévères nécessitant des transfusions itératives permettra de discuter la réalisation d’un sexe fœtal sur sang maternel et d’un éventuel diagnostic prénatal.1
DIAGNOSTICS DIFFÉRENTIELS
Votre texte ici
ÉTIOLOGIE
Votre texte ici
COMPLICATIONS
Votre texte ici
PRISE EN CHARGE THÉRAPEUTIQUE
A) Ictère néonatal
La prise en charge de l’ictère néonatal a pour but de prévenir et de corriger rapidement l’hyperbilirubinémie sévère afin d’éviter tout risque neurologique. Toutes les recommandations publiées promeuvent une approche basée sur trois étapes:1 1 1 1
- Dépistage systématique de l’ictère néonatal : intégrant d’une part les risques de sous-évaluation par le dépistage uniquement visuel en particulier sur les peaux foncées et d’autre part les facteurs cliniques de risque d’hyperbilirubinémie sévère (ictère avant H24, âge gestationnel <38 SA, origine familiale : Afrique, Antilles et Asie, et allaitement maternel inefficace). La bilirubinémie sera quantifiée de façon invasive (dosage sanguin) ou non invasive (réflectomètre transcutané). Le résultat sera analysé par comparaison avec les valeurs de référence de bilirubinémie pour l’âge post-natal en heure pour décider de la qualité (rythme et modalités) de la surveillance et/ou du traitement à mettre en place. Il est important de souligner que la gravité de l’hyperbilirubinémie est facilement sous-estimée par l’appréciation visuelle de l’intensité de l’ictère surtout sur les peaux foncées.
- Arsenal thérapeutique efficace mis en place rapidement : photothérapie (active l’élimination de la bilirubine par interaction entre la lumière et la peau), exsanguino-transfusion (épure le sang circulant de la bilirubine), agents pharmacologiques (interfèrent avec une des étapes du métabolisme de la bilirubine). L’hyperbilirubinémie par hémolyse secondaire au déficit en G6PD est une urgence thérapeutique car elle progresse très rapidement.
- Organisation d’un suivi individuel adapté intégrant l’évolutivité et la physiopathologie de l’ictère sur les premiers 15 jours de vie : le déficit en G6PD devient le plus souvent symptomatique à J4- J5 sur un mode aigu, donc après la sortie de maternité. Cette organisation repose sur la transmission des informations sur l’ictère durant le séjour en maternité (carnet de santé ou fiche de transmission), l’information des parents, la connaissance des signes d’hyperbilirubinémie sévère par les professionnels (enfant inconsolable ou au contraire somnolent, cri suraigu, cou en hyperextension, rejet de la tête en arrière représentent des urgences thérapeutiques) et les interactions étroites entre maternité et réseau d’aval (consultations de suivi sur la maternité, réseau d’accompagnement de retour à domicile, HAD). Enfin, si la surveillance risque de ne pas être optimale, en cas d'ictère précoce et sévère, la sortie est retardée.
La mise en place d’un dépistage néonatal systématique du déficit en G6PD reste un sujet de débat dans de nombreux pays et il est essentiellement organisé au seul échelon régional ou local.1 1
B) Prise en charge de l’hémolyse aiguë
- Le traitement est symptomatique et comporte l’arrêt de l’exposition à l’agent déclenchant (principalement le médicament causal) et une hydratation.
- Une transfusion de concentrés de globules rouges peut être indiquée selon la tolérance clinique et le degré d’anémie, l’hémoglobinurie témoignant d’une hémolyse toujours active.
- Une insuffisance rénale aiguë en règle réversible peut compliquer une hémolyse sévère et nécessiter une épuration extra-rénale.
C) Prise en charge et traitement de l’anémie hémolytique chronique des patients atteints d’un déficit de classe 1
- La prise en charge des patients atteints de déficit de classe 1 comporte des transfusions de concentrés de globules rouges, prescrites en cas d’aggravation de l’anémie chronique ou à un rythme régulier si l’anémie chronique est très sévère. Les transfusions sont phénotypées dans les systèmes Rh et Kell.
- Un traitement chélateur du fer se discute après 10-20 transfusions ou quand la concentration en fer hépatique déterminée par IRM atteint 125 micromoles/g de foie sec. La déféroxamine ou le déférasirox ont été administrés aux sujets déficitaires en G6PD.
- La supplémentation en acide folique (vitamine B9) est recommandée, celle en tocophérol (vitamine E) peut être proposée.
- La splénectomie peut être discutée en cas d’anémie sévère nécessitant des transfusions régulières mais est inconstamment efficace. Le suivi des patients ayant subi une splénectomie doit obéir aux règles habituelles dans ce cas.
- La cholécystectomie est indiquée en cas de calculs biliaires, lorsqu’ils sont symptomatiques ou dans le même temps qu’une splénectomie.
- Les vaccinations sont proposées selon le calendrier vaccinal de l’enfant y compris celles contre la grippe et l’hépatite B.
- Les accidents hémolytiques secondaires à un stress oxydatif surviennent également chez les patients avec déficit de classe 1 aggravant l’anémie chronique déjà présente. Les règles générales d’éviction médicamenteuse et alimentaire sont donc strictement à appliquer.
ÉVOLUTION/PRONOSTIC
Votre texte ici
PRÉVENTION
Les accidents hémolytiques aigus déclenchés par l’exposition à un agent oxydant peuvent facilement être prévenus par des mesures simples d’éviction des agents. Ces mesures doivent être appliquées tout au long de la vie, chez toute personne déficitaire en G6PD quels que soient ses antécédents et le type de mutation dont elle est atteinte ainsi que par les mères enceintes ou allaitant des nouveau-nés et nourrissons potentiellement déficitaires. Les personnes ayant déjà été exposées à des produits contre-indiqués sans que cela n'ait entraîné de conséquences demeurent à risque d’accident hémolytique au cours d’une prochaine exposition à ces mêmes produits.
A) Eviction médicamenteuse
- En 2014, l'ANSM a publié la mise à jour du document « médicaments et déficit en G6PD » qui comporte la liste recensant les médicaments susceptibles de provoquer une hémolyse chez les patients atteints de déficit en G6PD (http://ansm.sante.fr/g6pd). Cette liste est établie à partir des données de la littérature et de pharmacovigilance. A noter qu’il n'existe pas de tests in vivo/vitro permettant d’affirmer formellement le caractère hémolysant d’une molécule. Cette liste fait actuellement référence et il est recommandé aux patients de s'en munir pour toute consultation médicale. Elle établit 3 niveaux de précaution en séparant des médicaments contre-indiqués, des médicaments déconseillés, et des médicaments dont la dose maximale doit être respectée. Il est important que les professionnels de santé déclarent les accidents hémolytiques à leur Centre Régional de Pharmacovigilance (CRPV), et ce, même si le médicament mis en cause est déjà mentionné dans la liste comme déclencheur potentiel d'hémolyse (déclaration d'effet indésirable sur le site de l'ANSM)
B) Règles hygiéno-diététiques
- L’ingestion de fève (vicia faba ou féverole) provoque des accidents hémolytiques chez les personnes atteintes de déficit en G6PD. C’est pour cette raison que le déficit en G6PD est aussi appelé favisme. Il est donc formellement déconseillé de consommer des fèves qu'elles soient crues ou cuites, fraîches ou surgelées. On notera que dans certains pays comme l’Egypte ou la Chine, la fève fait partie de l’alimentation de base. L'hémolyse peut être variable d'un sujet à l'autre ou chez un même sujet, c'est pourquoi la contre-indication des fèves est valable pour tous les déficitaires quel que soit leur type de déficit. Quelques rares cas non prouvés de favisme ont été imputés à l’inhalation de pollen de fèves.
- Hormis les fèves, tous les autres fruits et légumes sont autorisés chez les sujets déficitaires en G6PD. Seuls les apports élevés de vitamine C par le biais de compléments alimentaires sont à éviter de façon à ne pas dépasser la dose de 1g/jour. La quinine (contenue dans certaines boissons toniques, énergisantes ou apéritives) peut provoquer des hémolyses aiguës en cas de déficit en G6PD.
- Des cas d’hémolyse ont été rapportés suite à une exposition au henné, cutanée chez le petit enfant ou digestive ainsi bien que moins bien documentés après l'ingestion de la plante médicinale acalypha indica. Il est recommandé de ne pas consommer de préparations à base d’extraits de plantes ou de vitamines dont la composition est inconnue. L’exposition à quelques rares autres produits comme les dérivés nitrés (contenu dans les « poppers ») est responsable de cas d’hémolyse aigue et de methémoglobinémie. Le naphtalène notamment contenu dans les boules anti-mites peut également provoquer par contact, inhalation, ingestion accidentelle des complications hémolytiques chez les nourrissons en particulier.
C) Éducation des patients
Le patient doit être clairement informé des recommandations à suivre pour éviter les accidents d'hémolyse aiguë. Il n'y a pas de gradation dans la prévention : les mêmes règles s'appliquent à tout sujet déficitaire quel que soit l’importance et le type du déficit enzymatique. Il est à noter que plusieurs listes erronées d'aliments contre-indiqués circulent toujours, notamment sur internet.
Les recommandations à suivre sont :
- se munir de la liste de médicaments émise par l'ANSM (2014) pour toute consultation médicale de façon à vérifier la dangerosité potentielle du médicament prescrit. L’automédication est fortement déconseillée.
- respecter strictement les doses recommandées de vitamine C, d’acide acétylsalicylique et de paracétamol
- respecter les règles hygiéno-diététiques citées au paragraphe précédent en particulier de ne pas consommer de fèves
- faire établir sa carte de soins et d'urgence pour déficit en G6PD, commandée et remplie par le médecin traitant ou le spécialiste (carte disponible sur demande du médecin depuis 2015)
- pour les enfants, informer l'entourage et suivre les recommandations d'accueil en collectivité
D) Prévention chez les hétérozygotes de sexe féminin
- Les résultats des dosages enzymatiques sont très variables chez les femmes hétérozygotes. Ils sont le plus souvent à la limite inférieure de la normale ou modérément abaissés. Le risque d’hémolyse est en partie dépendant de la valeur de l’activité enzymatique et de l’importance de l’exposition chimique en cause.
- Si l’activité est franchement basse (femmes hétérozygotes avec un biais d’inactivation en faveur de l’X porteur de la mutation) les mêmes précautions que celles établies pour l’homme déficitaire ou les rares cas féminins homozygotes sont préconisées.
- Si l’activité est normale, même si l’hétérozygotie n’est pas exclue par le seul dosage, les précautions particulières ne sont pas obligatoires sauf en cas de grossesse ou d’allaitement.
- En effet, toutes les femmes portant ou allaitant un enfant potentiellement déficitaire doivent respecter les contre-indications médicamenteuses et alimentaires : des cas d’hémolyse chez le nouveau–né ou le nourrisson déficitaires ont été rapportés dans le contexte d’une exposition maternelle aux agents oxydants en particulier via l’allaitement.
SURVEILLANCE
A) Suivi clinique et para-clinique des sujets déficitaires
1) Suivi des patients déficitaires de classe 1 avec anémie chronique
- Il est similaire à celui de tout patient atteint d’anémie hémolytique chronique :
- Il comporte sur le plan clinique la surveillance de la croissance staturo-pondérale et du développement pubertaire chez l’enfant ainsi que de l’importance de la splénomégalie.
- Sur le plan biologique le taux d’Hb, des réticulocytes, de la bilirubine, des LDH sont suivis. La surveillance biologique d’une possible surcharge en fer associe ferritine sérique et coefficient de saturation de la transferrine.
- La surveillance annuelle de la sérologie du parvovirus B19 si le patient n’est pas immunisé et le contrôle du taux vaccinal d’AC-anti-HbS sont proposés.
- Le suivi radiologique comporte le dépistage des lithiases biliaires dès la petite enfance par l’échographie abdominale en règle tous les 1 à 2 ans et celui d’une éventuelle surcharge en fer (IRM hépatique et cardiaque).
- L’échographie cardiaque avec dépistage de l’HTAP est préconisée dans le cadre de l’anémie hémolytique chronique, tout particulièrement chez les patients splénectomisés. La transition des soins pédiatriques vers la prise en charge en médecine adulte sera planifiée et organisée.
2) Suivi pour les patients porteurs d’un déficit de classe 2 et 3
- La majorité des sujets déficitaires restera asymptomatique tout au long de l’existence. Pour les déficitaires de classe 2-3, il n’existe à l’état basal ni splénomégalie, ni anémie, ni ictère.
- Il n’y a pas de traitement ou de surveillance au long cours à mettre en place.
- L’éducation permet par la connaissance des facteurs déclenchant, de prévenir en grande partie la survenue des accidents hémolytiques. Si l’âge au diagnostic est pédiatrique, une seconde consultation ciblée sur le déficit est recommandée afin de réitérer les conseils de prévention et l’information sur les signes de l’hémolyse : à la famille au moment de la scolarisation de l’enfant en cas de diagnostic néonatal et quand le sujet devient lui-même l’interlocuteur direct de la consultation (adolescent ou adulte). Cette nouvelle consultation permet de faire un point sur les documents remis et leur éventuelle mise à jour, d’élargir si besoin le bilan familial et de réexpliquer la transmission de la maladie. Si le médecin traitant reste l’acteur principal de l’éviction des médicaments oxydants, le déficit en G6PD doit être systématiquement signalé à tout nouvel intervenant médical.
- Certaines infections particulières ont été associées à la survenue d’une hémolyse aiguë. De nombreux cas d’hépatite virale aiguë sévère en particulier d’hépatite A ont été rapportés chez des sujets méditerranéens et asiatiques déficitaires en G6PD. Dans ce contexte, il est rappelé qu’en France la vaccination contre l’hépatite A est recommandée chez les adultes et enfants de plus d’un an susceptibles de séjourner dans un pays de haute endémicité. Même si le déficit en G6PD confère un certain niveau de résistance à l’infection au paludisme à l’échelle des populations, la prévention du paludisme reste formellement indiquée à l’échelle individuelle.
- Il n’a pas été décrit pour les patients déficitaires de classe 2-3 d’impact négatif du déficit sur l’espérance de vie ni de conséquences sur leur activité physique.
- Une majoration du risque de lithiase biliaire a été rapportée chez les sujets déficitaires Med(B-) ne justifiant cependant pas de dépistage échographique systématique.
- Le déficit en G6PD n’est pas dépisté de manière systématique en France, ni chez les nouveau-nés, ni chez les donneurs de sang, ni chez les militaires.
- Chez les donneurs de sang en France, si le déficit en G6PD est connu, il constitue une contre-indication au don de sang.
B) Hospitalisations et admissions en urgence
- Le patient et son entourage seront informés des principaux signes cliniques survenant lors d’un accident hémolytique (urines couleur porto, pâleur, asthénie, essoufflement) lors de l’annonce du diagnostic puis à nouveau par le médecin traitant au cours du suivi. La survenue de ces signes nécessite une consultation médicale en urgence. Un épisode d’hémolyse aiguë peut mettre en jeu le pronostic vital du patient de par la sévérité et la rapidité d’installation de l’anémie.
- Le déficit en G6PD sera signalé à chaque admission en urgence quel qu’en soit le motif par le patient et par le médecin adressant son patient. La carte de soins sera présentée. Egalement lors de toute consultation d’anesthésie en particulier pour une chirurgie à risque d’infection ou d’hypoxie, le déficit en G6PD doit être signalé.
CAS PARTICULIERS
Votre texte ici
THÉRAPIES FUTURES
Votre texte ici