Neuroblastome

INTRODUCTION/GÉNÉRALITÉ
- Le neuroblastome est la tumeur solide extra-crânienne la plus fréquente de la jeune enfance.
- Il s'agit d'une tumeur embryonnaire dérivée du système nerveux sympathique embryonnaire correspondant aux cellules de la crête neurale qui forment la médullosurrénale et les ganglions sympathiques.
- La grande hétérogénéité clinique, avec un spectre allant de la possibilité d'une régression spontanée jusqu'à une progression menaçante malgré tout traitement, ainsi que la grande hétérogénéité biologique ont suscité d'intenses recherches sur les facteurs pronostiques cliniques, radiologiques et biologiques et l'identification de nouvelles approches thérapeutiques.1
HISTORIQUE
Votre texte ici
ANATOMOPATHOLOGIE
Une analyse pathologique soit d'une pièce d'exérèse chirurgicale soit d'une biopsie chirurgicale ou percutanée, obtenue lors du diagnostic, permet une confirmation du diagnostic histologique et une classification plus précise selon les critères de l'INPC.
Les tumeurs neuroblastiques périphériques présentent différents degrés de différenciation, distinguant ainsi :
- le ganglioneurome (les cellules tumorales présentent une différenciation terminale) ;
- le ganglioneuroblastome (certaines cellules tumorales présentent la différenciation neuronale, d'autres correspondent aux cellules ganglionnaires) ;
- le neuroblastome (principalement des petites cellules tumorales immatures, rondes).
La prise en compte de la composante des cellules schwanniennes stromales permet de classer les tumeurs neuroblastiques en quatre catégories :
- neuroblastome (stroma pauvre) ;
- ganglioneuroblastome mélangé (stroma riche) ;
- ganglioneuroblastome nodulaire ;
- ganglioneurome (stroma dominant).
L'INPC définit une classification histopronostique en prenant en compte :
- ces catégories ;
- le degré de différenciation (en voie de différenciation, peu différencié, indifférencié) ;
- l'indice de mitose et karyorrexis (MKI) des cellules tumorales ;
- l'âge au diagnostic.
ÉPIDÉMIOLOGIE
- La prévalence du neuroblastome est estimée à 1 cas par 8 000–10 000 naissances, avec une incidence annuelle de 1 cas par 100 000 enfants, soit environ 120–150 nouveaux cas par an en France.
- Alors qu'il s'agit le plus souvent d'une pathologie sporadique, des rares cas familiaux ont été décrits.
- L'âge médian au diagnostic est de 18 mois, avec 40 % des patients diagnostiqués avant l'âge de 1 an, et 90 % avant l'âge de 5 ans. L'âge au diagnostic est un facteur pronostique important, les patients de moins de 18 mois ayant en général un meilleur pronostic.
- Un neuroblastome chez les adolescents et les adultes est très rare, grave et montre généralement une évolution clinique plus indolente.
génétique
L'étude de la biologie moléculaire des tumeurs neuroblastique apporte des informations pronostiques supplémentaires importantes.
L'oncogène MYCN, localisé au niveau du chromosome 2p24.3, peut être amplifié au niveau tumoral, avec présence de dizaines de copies de ce gène. Ce gène joue un rôle important dans l'oncogenèse de ces tumeurs et l'amplification, retrouvée dans 20 à 25 % des cas, représente un facteur pronostique péjoratif important même en cas d'une maladie localisée ou chez les nourrissons. Le statut génomique du gène MYCN est déterminé sur un prélèvement tumoral en utilisant la FISH ou plus rarement d'autres techniques moléculaires. D'autres altérations génétiques somatiques concernent des variations en nombre de copies des chromosomes, déterminé le plus souvent par une analyse en biologie moléculaire de type CGH-a ou SNP-array et permettant une analyse globale du génome tumoral :
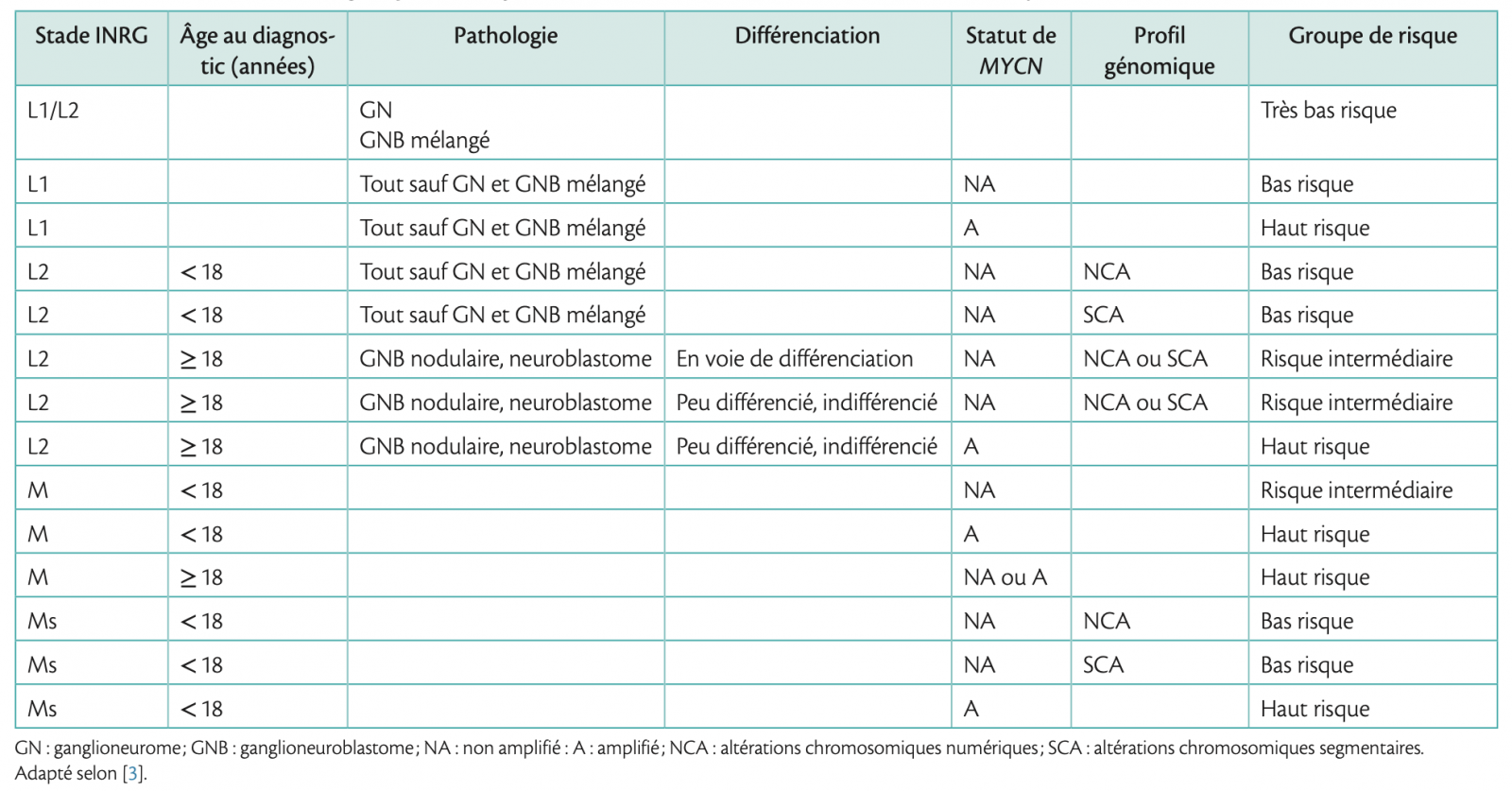
- une perte ou gain de chromosomes entiers correspond à une variation de la ploïdie ; ce profil génomique, aussi appelé «numérique» (NCA : Numerical Chromosome Alteration), est associé à un pronostic favorable (figure ci dessous) ;
-
d'autres altérations portent sur des bras chromosomiques, telles qu'une délétion des bras chromosomiques 1p, 3p, 4p, 6q ou 11q, ou un gain des bras chromosomiques 1q, 2p ou 17q. Ces altérations, appelées «segmentaires» (SCA : Segmental Chromosome Alterations), sont associées à un pronostic plus défavorable.1
D'autres altérations génétiques peuvent être retouvées au niveau des cel- lules neuroblastiques. L'utilisation des technologies récentes de séquençage haut débit a permis la mise en évidence de mutation activatrice du gène ALK dans 8 à 10 % des cas. D'autres mutations de la voie de signalisation RAS-MAPK, de TERT ou des altérations impliquant des gènes de remodelage de la chromatine ont été retrouvées, et de nombreuses études portent actuellement sur la définition de leurs rôles dans l'oncogenèse, leurs rôles pronostiques et surtout leurs rôles prédictifs d'une possible réponse à des traitements ciblés.
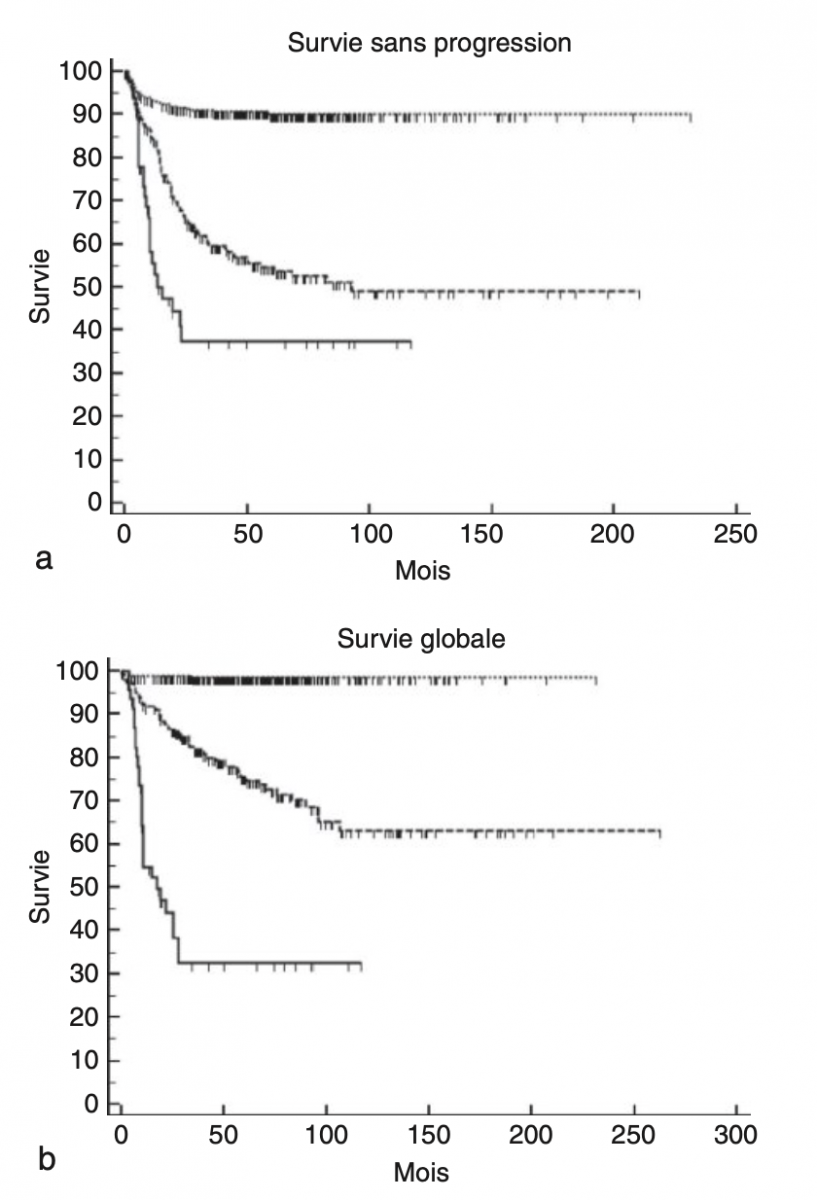
Pour les patients avec un profil tumoral génomique montrant uniquement des altérations chromosomiques numériques (NCA, n = 332), la survie sans progression et la survie gobale à 5 ans sont de 90 (± 1,7 %) et de 98 % (± 0,6 %) respectivement. Pour les patients avec un profil tumoral génomique montrant des altérations chromosomiques segmentaires (SCA, n = 201), la survie sans progression à 5 ans est de 55 % (± 3,3 %). Pour les patients dont la tumeur montre une amplification de MYCN (MNA, n = 46), la survie sans progression et la survie gobale à 5 ans sont de 37 (± 8 %) et 32 % (± 7,5 %) respectivement.1
EXAMEN CLINIQUE
Les symptômes cliniques du neuroblastome sont liés à la localisation de la tumeur primitive et des sites métastatiques. Les tumeurs primitives peuvent survenir n'importe où le long du système nerveux sympathique, avec une origine surrénalienne dans plus de 50 % des cas, et une localisation abdominale dans 80 %. Des formes bilatérales peuvent survenir dans un contexte de prédisposition génétique.
Le diagnostic d'une maladie localisée, non métastatique, peut être fait dans le cadre d'une découverte fortuite, ou peut être associé à des signes cliniques en rapport avec la localisation tumorale :
- Les tumeurs de localisation abdominale peuvent être à l'origine d'une distension abdominale, de douleurs, d'une hypertension artérielle.
- Les tumeurs de localisation cervicale ou thoracique peuvent provoquer un syndrome de Horner (ptosis, myosis, enophtalmie, anhidrosis), ou des signes respiratoires compressifs.
- Les tumeurs de point de départ ganglionnaire sympathique paraspinal peuvent se développer le long des racines nerveuses et pénétrer au niveau foraminal, sous forme de tumeur en sablier, conduisant à une compression médullaire.
Une atteinte métastatique est détectée chez environ 50 % des patients au moment du diagnostic, avec le plus souvent une atteinte ganglionnaire, une atteinte médullaire ou osseuse, alors qu'une atteinte hépatique ou cutanée peut être observée en particulier chez les nourrissons. Une atteinte pulmonaire ou du système nerveux central est observée plus rarement au diagnostic et est associée à un pronostic défavorable. Une atteinte métastatique peut être à l'origine de signes généraux tels que douleurs, fièvre ou perte de poids, ou de signes plus spécifiques telle qu'une douleur osseuse localisée. Les métastases périorbitaires provoquent parfois des ecchymoses et hématomes périorbitaires (syndrome de Hutchinson). D'autres symptômes cliniques comme une détresse respiratoire, des troubles de la coagulation ou une insuffisance rénale surviennent plus rarement.
Le neuroblastome peut se développer dans le cadre de syndromes paranéoplasiques associés :
- Une hypersécrétion du peptide vaso-intestinal (VIP) peut conduire à une diarrhée aqueuse abondante.
- Le syndrome opso-myoclonique (SOM) est un syndrome neurologique rare caractérisé par l'association d'une opsoclonie (mouvements conjugués rapides, multidirectionnels des yeux), de myoclonies et une ataxie cérébelleuse, très probablement à la suite d'un processus auto-immun. Ce syndrome est observé chez 2 à 3 % des enfants atteints d'un neuroblastome, alors qu'un neuroblastome peut être détecté chez 50 à 80 % de tous les enfants atteints d'un SOM.
EXAMENS COMPLÉMENTAIRES
A) Biologie
- Une élévation de la sécrétion urinaire des catécholamines ou de leurs métabolites, évaluée sur la dopamine, l'HVA et/ou le VMA, peut être détectée chez 90 % des patients. Le profil des catécholamines urinaires reflète le degré de maturation cellulaire avec un taux de dopamine ou un ratio de HVA/VMA élevé associé à un pronostic défavorable. Rarement, une élévation de noradrénaline ou adrénaline conduit à une hypertension artérielle.
- Un taux élevé d'autres marqueurs sériques tels que la LDH ou NSE (Neuron-Specific Enolase) a été associé à un pronostic défavorable, mais ces marqueurs ne sont pas spécifiques.
B) Imagerie
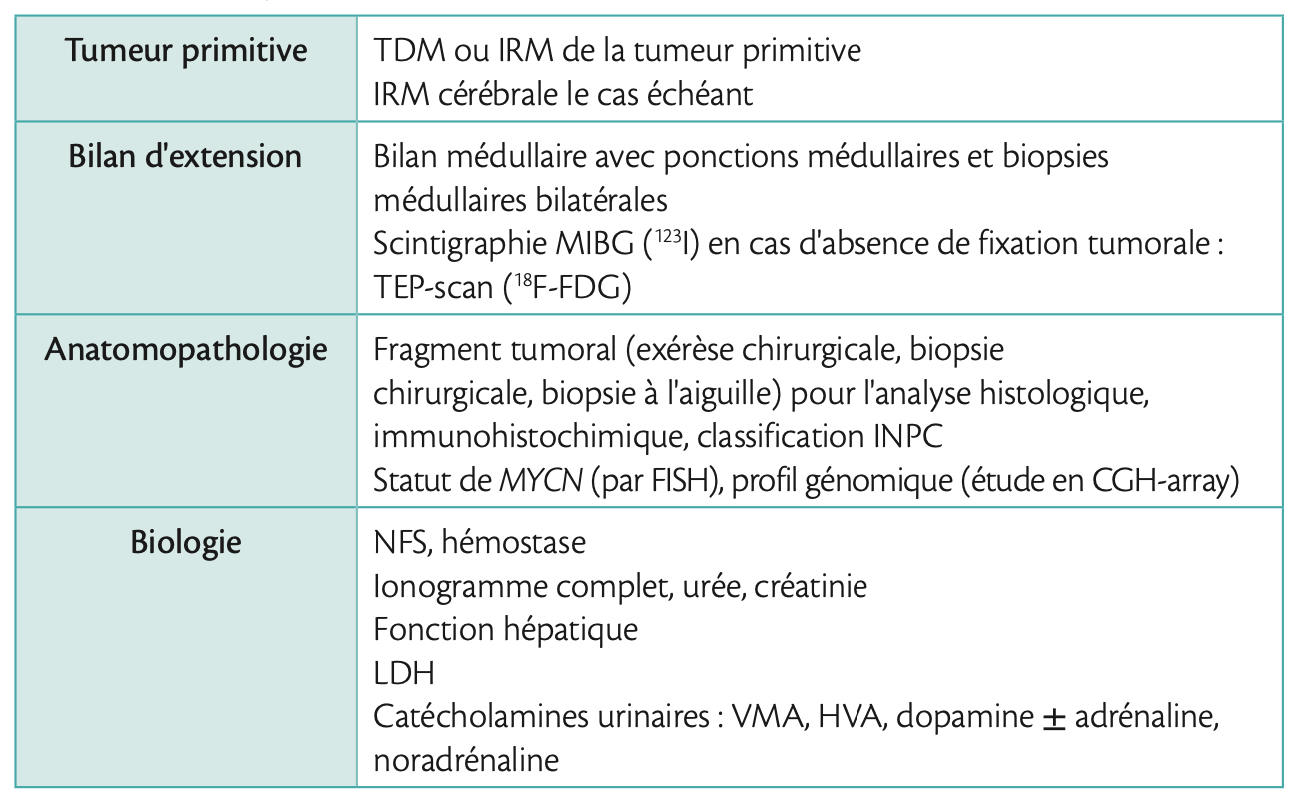
Le bilan diagnostique et d'extension initial comprend une évaluation radiologique de la tumeur primitive et une recherche des possibles métastases.
- Bien que l'échographie soit souvent le premier examen d'imagerie en raison de sa grande disponibilité et de son caractère non invasif, l'évaluation locale doit comporter soit une TDM, soit une IRM, avec une préférence pour l'IRM, en raison d'une résolution de contraste plus élevée et d'une absence d'exposition aux rayonnements ionisants, et ceci en dépit des temps d'acquisition plus longs et des besoins de sédation chez les jeunes enfants. Les tumeurs neuroblastiques sont souvent hétérogènes avec présence de calcifications et atteinte des ganglions lymphatiques régionaux. L'étude précise de l'extension locale permettra de rechercher des facteurs de risque définis en imagerie (IDRF) par rapport à une possible exérèse chirurgicale en prenant en compte une atteinte périvasculaire, l'infiltration des tissus et des organes adjacents (tels que les reins ou le foie), l'infiltration foraminale et de l'espace épidural du canal rachidien. Dans le cadre du système de stadage international (INRGSS, International Neuroblastoma Risk Group Staging System), l'étendue de la maladie locale est classée selon l'absence ou la présence d'IDRF, permettant une classification des tumeurs localisées en L1 (tumeur limitée à un compartiment du corps et de l'absence de tout IDRF) ou L2 (présence d'IDRF).1 1
- L'étendue de la maladie métastatique est évaluée par une scintigraphie de MIBG, en utilisant de préférence l'iode 123 (123I) plutôt que 131I en raison d'une exposition aux rayonnements ionisants plus faible, des images de meilleure qualité et d'une toxicité thyroïdienne moindre. Environ 90 % des neuroblastomes captent la MIBG. Différents scores (SIOPEN score, Curie score) ont été développés pour permettre une documentation semi-quantitative et une comparaison inter-individuelle des résultats.
- L'évaluation métastatique complète nécessite également un bilan médullaire avec en règle générale des ponctions médullaires et des biopsies médullaires bilatérales le plus souvent au niveau de la crête iliaque, avec un examen histologique et immuno-histochimique à la recherche de cellules neuroblastiques métastatiques.
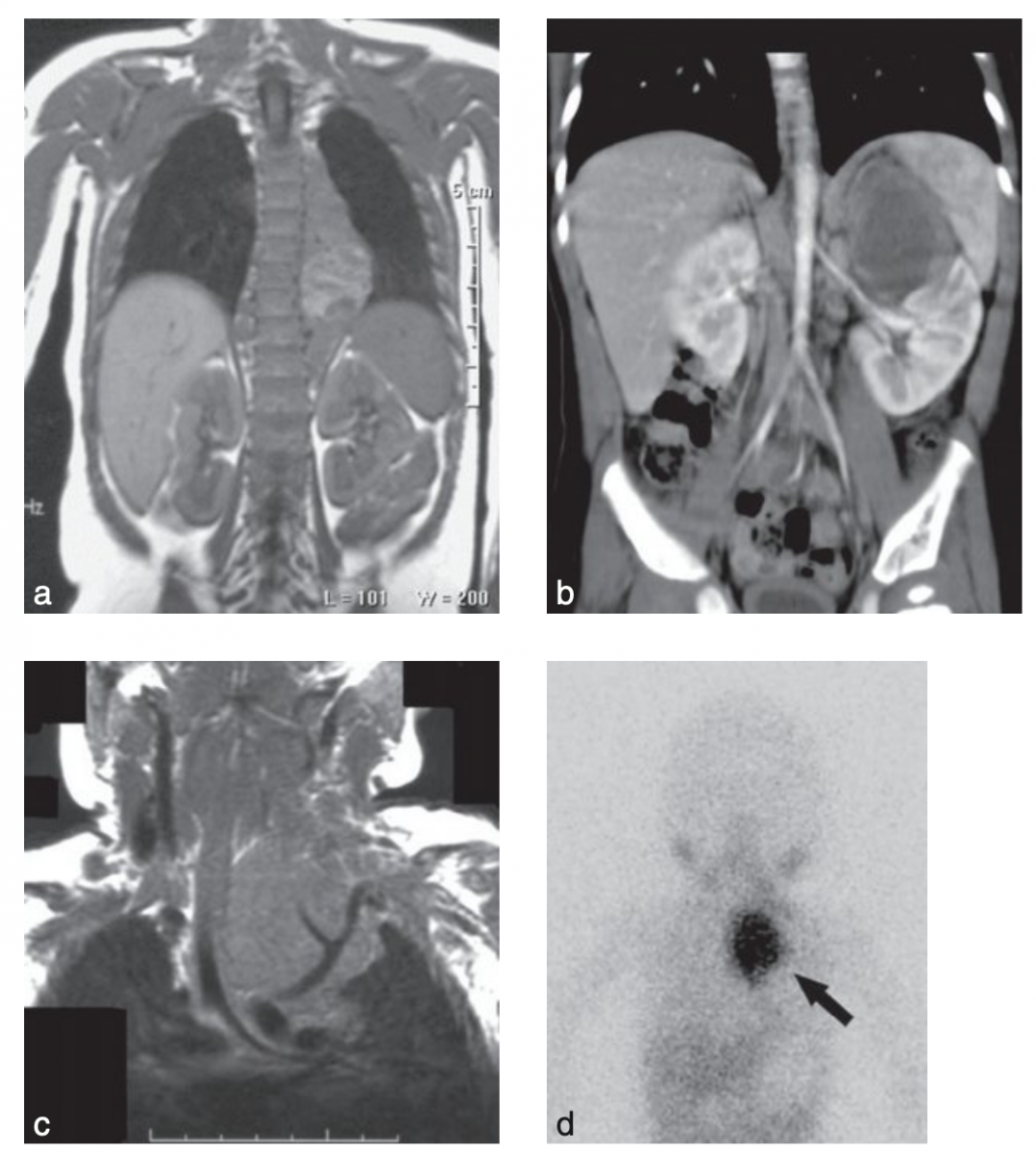
b. Neuroblastome de localisation surrénalienne gauche.
c. Neuroblastomede localisation médiatisnale supérieure.
d. Fixation intense locale à la MIBG sans fixation à distance.
Source : G. Schleiermacher.
C) Explorations initiales d'un neuroblastome.
DIAGNOSTICS DIFFÉRENTIELS
Votre texte ici
ÉTIOLOGIE
Votre texte ici
COMPLICATIONS
A) Compression médullaire
- Une compression médullaire survient chez environ 5–10 % de tous les patients atteints d'un neuroblastome, et constitue une urgence médicale absolue.
- Un traitement immédiat doit cibler les chances de récupération neurologique en cas de déficit neurologique.
- En plus du traitement symptomatique utilisant des corticoïdes à forte dose, les options de traitement incluent la chimiothérapie ou une intervention neurochirurgicale, soit par laminotomie soit par laminectomie.
- Bien que de nombreux investigateurs préfèrent une approche de chimiothérapie, des discussions multidisciplinaires détaillées sont nécessaires afin de permettre des décisions visant à diminuer la paraplégie ou d'autres séquelles.
B) SOM
- Le SOM est associé à des séquelles neurologiques, notamment neurocognitives, à moyen et long terme, malgré un pronostic oncologique favorable.
- En raison du mécanisme auto-immun sous-jacent, un traitement par corticothérapie à forte dose (bolus de dexaméthasone) est proposé.
- En cas de réponse insuffisante, ce traitement peut être associé à un traitement par cyclophosphamide, puis rituximab, ou si nécessaire des immunoglobulines polyvalentes, des agents immunomodulateurs ou plus rarement, dans les cas graves, la plasmaphérèse.
C) Sécrétion de VIP
- Une diarrhée sécrétoire survenant suite à une sécrétion de VIP nécessite souvent une réhydratation par voie intraveineuse
- Dans des rares cas un traitement par octréotide peut être nécessaire en cas de persistance des symptômes après résection de la tumeur.
PRISE EN CHARGE THÉRAPEUTIQUE
La prise en charge thérapeutique des patients atteints d'un neuroblastome dépend des groupes de risques et reflète l'hétérogénéité de cette pathologie, avec un spectre très large des possible prises en charge, d'une observation simple jusqu'au traitement multimodal intensif.

A) Neuroblastomes de bas risque et de risque intermédiaire
Près de la moitié des patients ont un neuroblastome de bas risque lors du diagnostic, comprenant des stades INRG L1, L2 et Ms avec les caractéristiques définies ci-dessus. Pour ces patients, les décisions thérapeutiques visent à fournir le minimum de traitement afin d'éviter au maximum toute séquelle, tout en conservant une excellente survie.
- Les nourrissons (< 1 an) avec une masse surrénalienne localisée peuvent probablement être observés en toute sécurité, même sans documentation histologique, sous réserve d'une étude du statut de MYCN sur le plasma.
- Pour les patients de plus de 1 an avec une maladie localisée qui semble être résécable en raison de l'absence d'IDRF (stade INRG L1) ou sur la base de l'évaluation chirurgicale, la tumeur doit être réséquée. En l'absence d'amplification de MYCN, une tumeur résiduelle n'est pas considérée comme un facteur de risque de rechute, avec une survie sans événement à 5 ans de plus de 90 % et une survie globale de 99 à 100 % pour ces patients.
- Pour les patients atteints d'un neuroblastome de bas risque, stade INRG L2 ou Ms (< 18 mois), la stratégie thérapeutique globale dépend des symptômes cliniques ainsi que des facteurs pronostiques plus précis, notamment du profil génomique. Pour ces patients, en l'absence de symptômes cliniques et en cas d'un profil géno- mique favorable, une observation simple peut être proposée en cas de stade Ms, et la faisabilité d'une telle approche est en cours d'étude dans le cadre d'un essai randomisé international.1 En présence de symptômes cliniques, ou en cas de facteurs pronostiques défavorables, notamment d'un profil génomique defavorable, un traitement par chimiothérapie est indiqué, en limitant le nombre de cycles à un minimum. Il n'y a pas d'indication de résection complète de la tumeur primitive en cas de persistance d'IDRF ni de radiothérapie pour ces patients.
- Pour les enfants ayant un neuroblastome de risque intermédiaire (stade INRG L2 MYCN non amplifié > 18 mois ou M < 18 mois), 2 à 8 cycles de chimiothérapie sont proposés, avec des combinaisons de chimiothérapie associant étoposide et carboplatine ainsi que vincristine, cyclophosphamide et doxorubicine. La résection chirurgicale de la tumeur primitive est effectuée lorsque cela est possible. En cas d'une tumeur de stade INRG L2 > 18 mois, en raison d'une survie globale à 5 ans inférieure à 60 %, une radiothérapie est également justifiée.
-
Dans l'avenir, il est prévisible que l'intensité du traitement des patients atteints d'un neuroblastome de risque intermédiaire soit adaptée en fonction de la réponse aux traitements et d'autres critères génétiques.1
B) Neuroblastomes de haut risque
- Ils concernent les patients atteints d'un neuroblastome stade INRG M chez les enfants de plus de 18 mois ou ceux dont la tumeur contient une amplification de MYCN.
- Bien que le taux de survie à 5 ans soit passé de moins de 15 % dans les années 1980 au taux actuel de 40 %, de nouvelles avancées thérapeutiques sont impératives.
L'approche actuelle intègre :
- chimiothérapie d'induction pour réduire la tumeur primitive et les métastases ;
- traitement local par chirurgie et radiothérapie ;
- consolidation avec chimiothérapie haute dose et réinjection de cellules- souches hématopoïétiques autologues (ASCT) ;
- traitement d'entretien en situation de maladie résiduelle avec un anticorps monoclonal anti-GD2 et une thérapie de différenciation par l'isotrétinoïne (acide rétinoïque).
1) Chimiothérapie d'induction
- Elle a pour objectif d'obtenir une rémission métastatique complète ou partielle. Les régimes actuels proposent des combinaisons associant étoposide, carboplatine, cisplatine, cyclophosphamide, vincristine, avec ou sans doxorubicine, permettant un taux de réponse complète et partielle de 70 à 80 % en fin d'induction. Une étude actuelle compare deux régimes d'induction, dont un propose des cures tous les 10 jours (Rapid COJEC) et l'autre un schéma de toutes les trois semaines (bras N7 modifié).1 1
2) Traitement local
- Une résection chirurgicale de la tumeur primitive est souvent difficile dans le neuroblastome à haut risque, même après chimiothérapie, en raison de l'englobement vasculaire fréquent. Une radiothérapie est administrée sur le lit tumoral à des doses de 21 à 36 Gy afin de diminuer le risque de récidive locale.
3) Chimiothérapie haute dose myéloablative avec autogreffe de cellules-souches périphériques
- L'introduction d'un traitement myéloablatif avec ASCT a permis une amélioration de l'EFS pour les patients atteints d'un neuroblastome de haut risque. Le recueil de cellules-souches hématopoïétiques est réalisé après obtention d'une rémission médullaire, par cytaphérèse. Initialement conçu en incluant une irradiation corporelle totale (TBI) pour traiter les foyers métastatiques, le traitement myéloablatif comporte aujourd'hui une chimiothérapie haute dose, ce qui permettra d'éviter ou de diminuer les effets tardifs d'une TBI comme les retards de croissance et les cancers secondaires. L'essai SIOPEN en cours a comparé un régime par melphalan-busulfan (BuMel) au régime de carboplatine, étoposide et melphalan (CEM), avec un avantage en termes d'EFS en faveur du bras BuMel. Ce traitement montre aussi un profil de toxi- cité à court terme plus favorable. Une possible complication est la maladie veino-occlusive (MVO), une micro-angiopathie se manifestant le plus souvent au niveau hépatique, mais parfois aussi au niveau pulmonaire. Le traitement par défibrotide peut être proposé en cas de MVO, en plus des traitements symptomatiques.
4) Traitement d'entretien en maladie résiduelle minime
- Malgré l'amélioration de l'EFS avec une chimiothérapie myélosuppressive suivie d'ASCT, 50 % des enfants vont présenter une rechute. L'ajout d'une molécule provoquant la différenciation des cellules neuroblastiques, l'acide rétinoïque, donné par voie orale, permet une amélioration de l'EFS.
- Le développement d'anticorps anti-GD2 monoclonaux a également conduit à une amélioration de l'EFS. Des études cherchent aujourd'hui à améliorer les modalités d'administration de ce traitement (perfusion de longue durée pour diminuer les effets secondaires notamment de type douleurs) et à étudier les possibles combinaisons (association avec des traitements immunomodulateurs comme les cytokines IL-2) en traitement d'entretien.
ÉVOLUTION/PRONOSTIC
- Le système de classification INRG a été conçu pour identifier les groupes de risque homogènes avant tout traitement pour permettre une stratification thérapeutique uniforme et une comparaison des essais cliniques menés par différents groupes coopératifs.1 Dans ce système, l'étendue de la maladie est déterminée par la présence ou l'absence d'IDRF et/ou la présence de métastases lors du diagnostic, avant tout traitement. Ainsi sont définis les stades localisées L1 et L2 et les stades métastatiques M et Ms (métastatique «spécial» du nourrisson avec des métastases cutanées, hépatiques et/ou médullaires mais pas osseuse), respectivement.
- D'autres facteurs pronostiques sont pris en compte dans cette classification, dont l'âge au moment du diagnostic (l'âge > 18 mois étant de pronsotic défavorable), la pathologie et les caractéristiques génétiques, y compris le statut de MYCN (la présence d'une amplification étant de pronostic défavorable) et le profil génomique (la présence d'altérations chromosomiques segmentaires étant de pronostic défavorable). Avec l'évolution des techniques en biologie moléculaire permettant des analyses encore plus détaillées au niveau de l'ADN et de l'ARN tumoral, il est probable que les groupes de risque seront précisés dans un avenir proche sur la base du profil moléculaire de la tumeur.
C) Prise en charge des rechutes
- En cas de rechute après traitement d'un neuroblastome de bas risque ou de risque intermédiaire, la stratégie de traitement du groupe de risque supérieur peut souvent être proposée. En cas de rechute de haut risque, la survie globale est très pauvre. Néanmoins, dans ces situations, de nouvelles combinaisons de chimiothérapie ont réussi à permettre une rémission partielle ou complète dans certains cas. Les traitements actuels reposent sur des combinaisons de chimiothérapie (en associant topotécan avec cyclophosphamide, irinotécan avec témozolomide, ou topotécan avec témozolomide). D'autres traitements de rattrapage utilisent une thérapie 131I-MIBG. Des combinaisons de traitements ciblés à la chimiothérapie sont actuellement à l'étude. Parmi les traitements ciblés, les inhibiteurs d'ALK (Anaplastic Lymphoma Kinase) peuvent être proposés aux 10–15 % de patients présentant une mutation de ce gène lors d'une rechute. D'autres thérapeutiques ciblées concernent les inhibiteurs d'aurora-kinase, les inhibiteurs de bromodomain, ou l'immunothérapie. Finalement, une stratégie plus simple avec étoposide per os est souvent proposée en phase palliative.
PRÉVENTION
Votre texte ici
SURVEILLANCE
Votre texte ici
CAS PARTICULIERS
Votre texte ici
THÉRAPIES FUTURES
- L'évolution des stratégies thérapeutiques pour les patients atteints d'un neuroblastome dépendra d'une amélioration de la compréhension de l'oncogenèse, sur le plan génétique et épigénétique, en prenant en compte le microenvironnement. tumoral ainsi que l'hôte lui-même. En intégrant ces nouvelles connaissances biologiques, la découverte de nouvelles cibles thérapeutiques et la compréhension de mécanismes de résistance, nous pourrons améliorer la prise en charge thérapeutique, en augmentant le taux de survie des patients avec un neuroblastome de haut risque tout en diminuant le risque de séquelles.